介绍
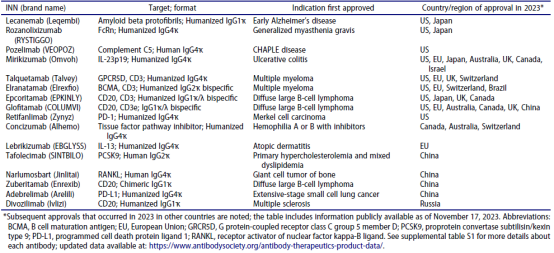
如表 1和下⽂摘要所述,截⾄2023年11⽉17⽇,共有16种新的抗体治疗产品
在2023年获得首次批准。还指出了2023年在其他国家进⾏的后续批准。在这16个产品中,⼤多数在美国获得批准(9/16),其中5个在中国和欧盟获得批准,4个在⽇本和加拿⼤获得批准。⽤于癌症的产品(8)和⽤于非癌症适应症的产品(8)数量相同。下面提供了产品关键详细信息的摘要(按表1中出现的顺序排列)。
表1. 2023年期间,商业赞助的单克隆抗体疗法在任何国家获得首次批准。
Lecanemab (Eisai Co., Ltd., Biogen, Inc.)
Lecanemab(BAN2401、LEQEMBI®)是一种⼈源化IgG1κ单克隆抗体,靶向可溶性和不溶性淀粉样蛋⽩β1-42 (Aβ) 聚集体。Lecanemab最初由BioArctic开发,通过合作协议授权给卫材(Eisai),从⽽实现联合开发。根据与Biogen的协议,Eisai和Biogen共同负责Lecanemab的商业化和推⼴。2023年1⽉6⽇,FDA加速批准Lecanemab-irmb⽤于治疗阿尔茨海默病(AD)。Lecanemab于2023年9⽉在⽇本获批上市。Lecanemab的上市申请正在欧盟、英国和中国接受审查。
FDA 的加速批准基于临床2期数据,该数据证明Lecanemab减少了⼤脑中 Aβ 斑块的积累,这是AD的一个明确的病理⽣理学特征。2023年7⽉6⽇,FDA 根据Eisai⼤型全球Charity AD临床试验(NCT03887455)的临床3期数据,将Lecanemab转为传统批准,该试验是一项多中⼼、随机、双盲、安慰剂对照、平⾏组研究招募了1,795 名AD患者。结果表明,与安慰剂相比,Lecanemab 在18个⽉时将临床痴呆评级总和下降减少了27%。Lecanemab的推荐剂量为每2周(Q2W)静脉输注10 mg/kg。患有轻度认知障碍或轻度痴呆阶段的患者应开始使⽤Lecanemab治疗,这些患者是在临床试验中评估治疗效果的⼈群。
Lecanemab的其他⼏项临床研究⽬前正在进⾏或招募参与者,包括一项临床2 期研究(NCT01767311)、针对患有AD致病基因突变个体的临床2/3期DIAN-TU-001(Tau NexGen;NCT05269394)研究,以及针对临床前AD和淀粉样蛋⽩升⾼或早期临床前AD和中间淀粉样蛋⽩患者的临床3 期AHEAD 3-45研究 (NCT04468659)。
Rozanolixizumab (UCB)
Rozanolixizumab (UCB7665, RYSTIGGO®) 是一种⼈源化IgG4ҡ单克隆抗体,可与新⽣⼉Fc受体(FcRn)结合。2023年6⽉27⽇,Rozanolixizumab-noli被FDA批准⽤于治疗抗⼄酰胆碱受体(AchR)或抗肌⾁特异性酪氨酸激酶(MuSK)抗体阳性的成⼈全⾝性重症肌⽆⼒(gMG)患者。MG是一种⾃⾝免疫性疾病,主要由⾃⾝抗体攻击位于神经肌⾁接头处的蛋⽩质引起,该蛋⽩质破坏了从神经到肌⾁的信号传输,导致肌⾁⽆⼒和疲劳。两种关键蛋⽩AchR和MuSK是⾃⾝抗体的靶标。通过与FcRn结合,Rozanolixizumab降低总IgG水平,从⽽导致AchR和MuSK⾃⾝抗体水平降低。
Rozanolixizumab 随后于2023年9⽉在⽇本获得批准,⽤于治疗对类固醇或其他免疫抑制剂反应不足的成⼈gMG患者。2023年11⽉9⽇,EMA药品委员会(CHMP)采纳了积极意⻅,并建议授予Rystiggo⽤于治疗gMG的上市授权。欧盟委员会的决定通常在意⻅通过后67天内发布。
Rozanolixizumab的批准得到了临床3期MycarinG研究(NCT03971422)数据的⽀持,该研究是一项多中⼼、随机、双盲、安慰剂对照研究。总共200名患者按 1:1:1随机接受体重分级剂量的Rozanolixizumab (n = 133),相当于
≈7 mg/kg (n = 66) 或
≈10 mg/ kg (n = 67),或安慰剂 (n = 67)。该研究达到了主要终点,第43天时,重症肌⽆⼒⽇常⽣活活动(MG-ADL)总分较基线变化具有统计学显着性差异,有利于Rozanolixizumab(Rozanolixizu-mab治疗组在任一剂量与安慰剂治疗组为-0.8分 (p < 0.001))。还达到了次要终点,即治疗组之间定量重症肌⽆⼒(QMG)从基线到第43天的变化。推荐剂量取决于患者的体重,每季度皮下输注,持续6周。50公⽄以下的患者接受的剂量为420毫克;50公⽄⾄100公⽄之间的患者用量560毫克,⽽体重100公⽄及以上的患者用量840毫克。
⽬前正在进⾏多项跨国临床3期和2期研究,或正在招募各种⾃⾝免疫性疾病的患者,包括gMG、髓磷脂少突胶质细胞糖蛋⽩抗体相关疾病、富含亮氨酸的神经胶质瘤灭活⾃⾝免疫性脑炎和严重纤维肌痛综合征。
Pozelimab (Regeneron Pharmaceuticals, Inc.)
Pozelimab (Veopox™, REGN3918) 是一种⼈源化IgG4κ mAb,靶向补体因⼦ 5(C5),有助于预防补体途径介导的疾病。Pozelimab 源⾃ Regeneron专有的 VelocImmune®转基因⼩⿏技术。FDA 授予Pozelimab罕⻅⼉科疾病称号,⽤于治疗CD55缺陷型蛋⽩丢失性肠病(CHAPLE),以及⽤于治疗CHAPLE和阵发性睡眠性⾎红蛋⽩尿(PNH)的孤⼉药。Pozelimab还获得了FDA的快速通道指定。
2023 年 8 ⽉ 18 ⽇,FDA 批准Pozelimab-bbfg⽤于治疗患有CHAPLE病的⼉童(>1 岁)和成⼈。该批准基于临床2/3期开放标签试验(NCT04209634) 的结果,该研究评估了Pozelimab对10名3⾄19岁(中位年龄8.5岁)接受 CHAPLE治疗的患者的疗效和安全性。患者在第1天接受单次负荷剂量30 mg/kg IV Pozelimab,然后每周接受基于体重的SC Pozelimab剂量。所有患者在12周内实现了⾎清⽩蛋⽩和⾎清IgG浓度的正常化,并且这种情况在⾄少72周的治疗中得以维持。常⻅的不良事件是上呼吸道感染、骨折、 荨⿇疹和脱发,Pozelimab的处⽅信息还包括关于接受补体抑制剂治疗的患者发⽣严重脑膜炎球菌感染⻛险的⿊框警告。临床试验中的所有患者在治疗前均接受了脑膜炎球菌疫苗接种,并接受抗菌药物作为预防性治疗。
Pozelimab还正在与Alnylam Pharmaceuticals, Inc.开发的siRNA C5抑制剂 Cemdisiran联合进⾏研究。临床3期研究(NCT05070858)正在评估Pozelimab 和Cemdisiran联合治疗对有症状的全⾝性症状成⼈患者的疗效和安全性。重症肌⽆⼒,以及临床3期NCT05744921和NCT05133531研究正在评估这种组合对PNH患者的作⽤。Regeneron预计在2025年或以后提交更多含有或不含 有Cemdisiran的Pozelimab的上市申请。
Mirikizumab (Eli Lilly)
Mirikizumab (LY3074828, Omvoh®) 是一种⼈源化抗IL -23p19 IgG4单克隆抗体,开发⽤于治疗溃疡性结肠炎(UC)和克罗恩病。引⼊了⼏个突变以稳定铰链区 (S228P)、减少FcγR和C1q结合 (F234A、L235A) 并消除C末端的异质性 (K447>del)。UC和克罗恩病都属于炎症性肠病(IBD)类别,其特征是消化道反复炎症。Mirikizumab以IL-23的p19亚基为靶点,抑制参与这些疾病发病机制的IL-23通路。
2023年3⽉27⽇,Mirikizumab在⽇本被批准⽤于对常规疗法或治疗反应不足的中重度UC患者的诱导和维持治疗。此次批准使Mirikizumab成为第一个⽤于该适应症的IL-23p19抑制剂。2023年5⽉26⽇,Mirikizumab在欧盟获得批准,⽤于治疗反应不充分、反应消失的中度⾄重度活动性UC成年患者对常规疗法或⽣物疗法或不耐受。随后,Mirikizumab于2023年9⽉在澳⼤利亚被批准⽤于治疗中度⾄重度活动性UC。2023年10⽉26⽇,Mirikizumab-mrkz被FDA批准⽤于治疗成⼈中度⾄重度活动性UC。Mirikizumab通过静脉输注给药在诱导治疗期间(每4周300毫克),在维持治疗期间通过⾃动注射器或注射器皮下注射(每4周200毫克)。
该批准基于LUCENT临床试验计划的结果。Mirikizumab在三项临床3期研究 LUCENT-1、LUCENT-2和LUCENT-3中进⾏了评估,其中包括UC患者。LUCENT-1 (NCT03518086) 是一项多中⼼、随机、双盲、安慰剂对照诱导研究,⼊组了1,100多名中重度活动性UC患者,先前失败的常规和/或⽣物疗法和/或JAK抑制剂,他们被随机分配⾄静脉注射300 mg Mirikizumab或安慰剂Q4W,为期12周。完成12周LUCENT-1诱导研究的患者 (n = 544) 在LUCENT-2 (NCT03524092)(一项多中⼼、随机)中被重新随机分组接受皮下注射
Mirikizumab (200 mg) 或安慰剂Q4W治疗另外40周,双盲,安慰剂对照维持研究。LUCENT-3 (NCT03519945) 是一项开放标签扩展研究,旨在评估 Mirikizumab对中度⾄重度活动性UC参与者的长期疗效和安全性。
对于 LUCENT-1和-2,主要结局指标分别是第12周和第40周临床缓解的参与者百分比。在LUCENT-1研究中,与服⽤安慰剂的患者(27.9%,n = 82/294,p < 0.001)相比,接受Mirikizumab治疗的患者获得了显着更⾼的临床缓解率 (45.5%,n = 395/868)。LUCENT-2研究结果显示,12周时获得临床缓解的患者中,63.6%(n = 91/143)通过Mirikizumab治疗能够在1年时维持临床缓解,⽽接受安慰剂的患者中这一比例仅为三分之一左右(36.9%,n = 24/65,p < 0.001)。两项研究均评估了使⽤紧急程度数字评定量表(0-10)测量的肠急迫性, 结果显示,与安慰剂相比,接受Mirikizumab治疗的患者在第12周和第52周在这项次要结果指标中实现了具有临床意义的改善的比例明显更⾼。
在临床3期 VIVID-1研究(NCT03926130)和长期扩展VIVID-2研究(NCT04232553)中,Mirikizumab也被评估为克罗恩病的治疗⽅法。Eli Lilly礼来公司最近宣布,VIVID-1研究达到了共同主要终点和所有主要次要终点,该研究在中度⾄重度活动性克罗恩病患者中评估了Mirikizumab与安慰剂或活性对照(Ustekinumab)的比较。根据研究结果,礼来公司计划向FDA提交 Mirikizumab治疗克罗恩病的BLA,并在2024年向其他监管机构提交。
Talquetamab (Janssen Research & Development, LLC)
Talquetamab (TALVEY™, JNJ-64407564) 是一种⼈源化 IgG4κ/λ双特异性T 细胞接合抗体,可与G蛋⽩偶联受体C类 5成员D (GPCR5D) 以及T细胞上的CD3结合。GPRC5D在骨髓瘤细胞表面⾼表达,在B细胞上表达极少。OmniAb的转基因⼩⿏和Genmab的双特异性双体技术被应⽤于Talquetamab单抗的开发中。
2023 年 8 ⽉ 9 ⽇,Talquetamab-tgvs 获得FDA加速批准,⽤于治疗复发性或难治性(RR)多发性骨髓瘤(MM)成年患者,这些患者之前接受过⾄少四种治疗,包括免疫调节剂剂、蛋⽩酶体抑制剂和抗CD38抗体。此外,2023年8⽉,Talquetamab获得欧盟委员会有条件上市授权,作为单一疗法⽤于治疗⾄少接受过三种治疗的RRMM成年患者。既往接受过治疗,包括免疫调节剂、蛋⽩酶体抑制剂和抗CD38抗体,并且在上次治疗中表现出疾病进展。
这些批准基于临床1/2期MonumenTAL-1研究(临床1期:NCT03399799,临床2期:NCT04634552)的积极结果,该研究最近在2023年美国临床肿瘤学会 (ASCO) 上公布,年会于2023年6⽉2-6⽇在芝加哥举⾏。临床1/2期MonumenTAL-1研究的患者接受0.8 mg/kg SC Talquetamab Q2W或0.4 mg/kg SC Talquetamab每周(QW)治疗,总体反应两种剂量的缓解率相似(Q2W总体缓解率为 73.6%(CI范围:63.0‒82.4),QW总体缓解率为 73%(CI范围:63.2‒81.4))。Q2W剂量的患者未达到中位缓解持续时间 (DOR),⽽QW剂量的患者的DOR为9.5个⽉(范围:6.7-13.3)。Q2W和QW剂量的12个⽉⽆进展⽣存(PFS)率分别为 54.4% 和34.9%。不良事件导致的停药率较低(Q2W为8%,QW为5%),最常⻅的不良事件是细胞因⼦释放综合征、味觉障碍和皮肤相关不良事件。导致治疗停止的不良反应主要是由于免疫效应细胞相关的神经毒性综合征或体重减轻。
⽬前还在评估Talquetamab与其他疗法的组合,例如⽤于治疗RRMM的抗 BCMA T细胞参与剂Teclistamab (TECVAYLI) (NCT04586426)、⽤于治疗MM的Teclistamab和抗CD38 Daratumumab (NCT04108195) ,一种⽤于RRMM的抗PD1抑制剂 (NCT05338775),⽤于治疗MM的carfilzomib、Daratumumab、lenalidomide和pomalidomide (NCT05050097),以及⽤于治疗难治性MM的Daratumumab、pomalidomide和地塞⽶松 (NCT05455320)。
Elranantamab (Pfizer Inc.)
Elranatamab (Elrexfio™, PF-06863135) 是一种⼈源化 IgG2κ双特异性T细胞接合抗体,靶向T细胞表面的CD3和B细胞成熟抗原 (BCMA),该抗原在骨髓瘤细胞表面⾼度表达。
2023年8⽉14⽇,FDA加速批准Elranantamab⽤于治疗RRMM成年患者,这些患者之前⾄少接受过四种疗法,包括免疫调节剂、蛋⽩酶体抑制剂和抗 CD38单克隆抗体。Elranatamab 随后于2023年9⽉在瑞⼠获得批准。FDA的审查是在Orbis项⽬下进⾏的,该项⽬是一个同时提交和审查肿瘤药物的框架,有可能加快国际合作伙伴的批准(瑞⼠、巴西、加拿⼤、澳⼤利亚和新加坡)。2023年12⽉,欧盟委员会授予Elranantamab有条件上市授权,⽤于治疗复发难治性多发性骨髓瘤(RRMM)成年患者,这些患者之前⾄少接受过三种治疗,包括蛋⽩酶体抑制剂、免疫调节剂和抗CD38抗体,并在最后一次治疗中显示出疾病进展。
这些批准是基于MagnetisMM-3临床2期(NCT04649359)的积极结果,其中患者在治疗的第一周期间给予两次启动剂量:12 mg和32 mg SC Elranatamab,然后每周给予76 mg SC Elranatamab。这项研究总共纳⼊了123名患者,他们对⾄少三种先前的治疗没有反应。61%(95% CI:51.8‒69.6%)的患者在中位随访14.7个⽉后获得了确认的客观缓解,其中35%的患者达到完全缓解或更好。六个周期后,患者具有部分缓解或更好持续⾄少2个⽉的患者改为Q2W给药间隔,⽽不是每周给药。在改⽤Q2W给药的患者中,80% 的患者在改⽤后⾄少6 个⽉仍保持反应。最常⻅的不良反应是细胞因⼦释放综合征、⾎液学相关事件和感染。
FDA 对Elranatamab的全面批准取决于验证性临床2期研究(MagnetisMM-5)的积极数据,该研究旨在测试Elranatamab单独治疗以及与Daratumumab联合治疗RRMM的效果,与标准治疗组合进⾏比较Daratumumab、Pomalidomide和地塞⽶松(NCT05020236)。该研究预计将包括多达854名患有RRMM的成年⼈,预计主要完成⽇期为2026年8 ⽉。
Epcoritamab (Genmab, AbbVie)
Epcoritamab(GEN3013,EPKINLY™)是一种针对CD20和CD3的IgG1k/λ T细胞接合剂。它由Genmab和AbbVie共同拥有,是使⽤Genmab的DuoBody®平台开发的。该⽅法利⽤受控的Fab臂交换,涉及两种IgG1抗体的⽣产、纯化和重组,每种抗体在第三恒定区(CH3)中包含一个匹配的点突变 (K409R; F405L)。选择这些突变是为了削弱亲本IgG1 mAb中的非共价CH3-CH3相互作⽤,并确保重轻 (HL) 链同⼆聚体解离的单向过程以及形成强烈有利的异⼆聚HL相互作⽤。2023年12⽉19⽇,FDA加速批准Epcoritamab -bysp⽤于治疗RR弥漫性⼤B细胞淋巴瘤(DLBCL)成⼈患者,未另有说明,包括惰性淋巴瘤引起的DLBCL和⾼级别B细胞淋巴瘤经过两次或多次全⾝治疗后。
EPCORE NHL-1(NCT03625037)是一项针对CD20+ DLBCL患者的开放标签、多队列、多中⼼、单臂试验,评估了Epcoritamab的有效性。在接受过⼤量治疗的RR DLBCL患者中,总体缓解率和完全缓解 (CR) 率分别为 61%(95% CI,53-69)和38%,估计中位缓解持续时间为15.6个⽉。Epcoritamab通过皮下注射以28天为一个周期进⾏给药,直⾄疾病进展或出现不可接受的毒性。处⽅信息附有⿊框警告,提示严重或危及⽣命的细胞因⼦释放综合征和危及⽣命或致命的免疫效应细胞相关神经毒性综合征。
AbbVie和Genmab⽬前正在进⾏一项临床3期试验(NCT04628494) 中评估Epcoritamab作为RR DLBCL患者的单一疗法。公司还在两项针对新诊断DLBCL (NCT05660967) 和RR滤泡性淋巴瘤 (FL) 患者的临床3期试验中评估EPKINLY联合⽅案(NCT05409066)。EPKINLY在美国和⽇本的商业责任将由AbbVie和Genmab 共同承担。AbbVie还负责进一步的全球商业化。
Glofitamab (Hoffmann-La Roche)
Glofitamab (RO7082859、CD20-TCB、RG6026、COLUMVITM)
是一种全长 IgG1κ/λ双特异性单克隆抗体,可同时结合恶性B细胞上的CD20和T细胞上的CD3。Glofitamab由Roche开发,采⽤CrossMab技术设计成 2:1 结构,其中通过柔性接头将第⼆个CD20臂与CD3ε结合臂融合,实现与CD20的⼆价结合。Glofitamab在Fc区还具有PG LALA突变,以消除与FcɣRs和C1q的结合。2023年3⽉25⽇,Glofitamab在加拿⼤获得批准(有条件),⽤于治疗未另⾏指定的RR DLBCL、FL引起的DLBCL或原发性纵隔B细胞淋巴瘤成⼈患者,这些患者已接受过两次或多次治疗更多线的全⾝治疗,并且没有资格接受或不能接受 CAR T细胞治疗或之前接受过CAR T细胞治疗。随后于2023年6⽉15⽇获得FDA加速批准, 2023年7⽉7⽇获得EM有条件上市授权以及其他国家的批准(表1)。
此次批准基于临床1/2 期NP30179研究(NCT03075696)的积极结果,该研究是一项开放标签、多中⼼、单臂试验,纳⼊了132名接受过两线或以上全⾝治疗后的RR DLBCL患者。接受固定疗程Glofitamab治疗的患者的总体缓解率为 56%(95% CI,47-65),43%达到完全缓解。首次反应的中位时间为42天。预计中位缓解持续时间为18.4个⽉。在第1周期第1天接受单剂量抗CD20 Obinutuzumab (1,000 mg) 后,通过静脉输注向患者施⽤Glofitamab,最多12 个周期(第1周期第8天2.5 mg、第15天10 mg ;以及后续每个21天周期的第一天30 mg)。研究中接受Glofitamab治疗的患者中最常⻅的不良事件是细胞因⼦释放综合征 (70%)
一项临床3期研究(STARGLO,NCT04408638)正在评估格Glofitamab联合gemcitabine + oxaliplatin (GemOx)与利妥昔单抗联合GemOx在复发/难治性 DLBCL患者中的疗效和安全性。该研究预计主要完成⽇期为2025年4⽉。
Retifanlimab (Incyte Corporation, Macrogenics, Zai Lab)
Retifanlimab (INCMGA00012、MGA012、ZL-1306,ZYNYZ™) 是一种⼈源化IgG4κ单克隆抗体,可与PD-1受体结合并阻断与其配体的相互作⽤。在2023年3⽉22⽇,FDA加速批准Retifanlimab -dlwr⽤于治疗转移性或复发性局部晚期默克尔细胞癌(MCC)成年患者。
此次批准基于POD1UM-201研究(NCT03599713)的疗效评估,该研究是一项开放标签、单组研究,对象为转移性或复发性局部晚期MCC患者,这些患者先前未接受过针对晚期疾病的全⾝治疗。主要终点是客观缓解率(ORR),由独⽴中央审查(ICR)使⽤实体瘤缓解评估标准(RECIST)v1.1确定。在接受Retifanlimab单药治疗的初治患者(n = 65)中,ORR为52% (95% CI, 40-65)。12名患者(18%)表现出完全缓解,22名患者(34%)出现部分缓解。26名 (76%)的缓解患者的缓解持续时间(DOR)为6个⽉或更长,21名患者的DOR为12个⽉或更长。Retifanlimab的推荐剂量为500 mg,每周四周静脉输注 30 分钟,直⾄疾病进展、出现不可接受的毒性或长达24个⽉。最常⻅(≥10%)的不良反应是疲劳、肌⾁骨骼疼痛、瘙痒、腹泻、皮疹、发热和恶⼼。
多项正在进⾏的临床试验正在评估Retifanlimab作为多种癌症类型的治疗⽅法,包括非⼩细胞肺癌(NSCLC)、阴茎鳞状细胞癌以及胰腺或壶腹腺鳞癌。
Concizumab (Novo Nordisk)
Concizumab(NNC172‒2021、NN7415、AlhemoTM)是一种⼈源化、铰链稳定(S228P 突变)IgG4κ单克隆抗体,可选择性结合组织因⼦途径抑制剂 (TFPI) 的Kunitz-2结构域。通过与TFPI结合,Concizumab降低其对激活的X因⼦(Xa 因⼦)(外源性凝⾎级联的重要组成部分)的抑制活性,并提⾼⾎栓形成的效率。2023年3⽉10⽇, Concizumab在加拿⼤获得批准,⽤于治疗患有B型⾎友病的青少年和成⼈患者(12岁或以上),这些患者患有凝⾎因⼦IX抑制剂,需要常规预防以预防或减少发⽣⾎友病的频率的出⾎事件。Concizumab随后分别于2023年7⽉和2023年8⽉在澳⼤利亚和瑞⼠获得批准。Concizumab的上市申请正在欧盟、美国和⽇本接受监管审查。2023年4⽉,诺和诺德宣布,他们收到了FDA的完整回复信,要求提供与患者监测和剂量以及生产工艺相关的更多信息。
Concizumab使⽤每⽇一次使⽤单⼈的预装多剂量笔皮下注射。Concizumab的推荐给药⽅案包括第1天的负荷剂量为1 mg/kg,随后在第2天和随后的⼏天每⽇一次剂量为0.20 mg/kg,直⾄设定个体维持剂量。维持剂量为根据开始治疗后 4周测量的Concizumab给药前⾎浆浓度确定。
Concizumab的疗效和安全性在临床3 期 explorer7试验(NCT04083781)中进⾏了评估。共有 133 名患者⼊组,其中52名随机接受旁路药物按需治疗(第 1组)或Concizumab预防治疗(第2组)。另外81 名非随机患者接受了 Concizumab治疗以评估总体安全性。该研究因与Concizumab治疗相关的⾎栓栓塞事件的发⽣⽽暂停,后来通过调整剂量⽅案恢复。研究暂停后获得的疗效结果表明,与接受旁路药物按需治疗的患者相比,接受Concizumab治疗的患者的⾃发性和创伤性出⾎减少了86%。在Concizumab预防组(n = 33),估计平均年化出⾎率(主要终点)较低,为1.7 (95% CI, 1.01‒2.87),⽽对照组为11.8 (95% CI, 7.03‒19.86) (n = 19)。
Concizumab 还在两项临床3期试验中进⾏评估explorer8(NCT04082429)⽤于治疗12岁以上患有A型和B型⾎友病的男性青少年和成⼈,⽆抑制剂;explorer10(NCT05135559)⽤于治疗12岁以下患有A型和B型⾎友病的男性⼉童和男性任何年龄的A型和B型⾎友病患者,⽆论是否有抑制剂。
Lebrikizumab (Almirall S.A., Eli Lilly and Company)
Lebrikizumab(Ebglyss)是一种⼈源化、铰链稳定(S228P突变)IgG4κ抗体,以IL-13为靶点,IL-13是促炎反应的关键介质,可增强神经元对特应性皮炎中持续瘙痒刺激的反应。Lebrikizumab最初由Roche集团成员F. Hoffmann-La Roche Ltd和Genentech, Inc.开发。2017年,Dermira(于2020年被礼来公司收购)获得了Lebrikizumab的专有权利,⽤于开发和商业化其治疗特应性皮炎和所有其他适应症。Roche保留某些权利,包括开发和推⼴治疗间质性肺疾病的Lebrikizumab的。Almirall于2019年获得了在欧洲开发和商业化 Lebrikizumab治疗特应性皮炎等某些适应症的专有权利。
2023年11⽉16⽇,欧盟委员会批准Lebrikizumab ⽤于治疗患有中度⾄重度特应性皮炎(AD)的成⼈和青少年患者(12岁及以上,体重⾄少40公⽄)。礼来公司(Lilly)于2022年第三季度向FDA 提交了Lebrikizumab(一种治疗特应性皮炎的药物)的BLA。FDA随后发布了一封完整的回应信,指出礼来公司计划解决的缺陷。
欧盟的批准基于三项临床3 期试验的结果,这些试验评估了Lebrikizumab在患有特应性皮炎的成⼈和12 岁以上青少年中的安全 性和有效性。Advocate 1(NCT04146363)和Advocate 2(NCT04178967)是随机、双盲、安慰剂对照、平⾏组研究,其中中度⾄重度特应性皮炎患者接受初始剂量500 mg
Lebrikizumab,随后接受250 mg Lebrikizumab Q2W 或安慰剂,治疗期为16周。16周后,对Lebrikizumab产⽣临床反应的患者被重新随机分配接受 Lebrikizumab Q2W 或Q4W或安慰剂,持续36周。主要终点是透明或⼏乎透明(分别为0 或1)皮肤的研究者总体评估(IGA)评分,较基线减少⾄少2分,湿疹面积和严重程度指数 (EASI) 减少⾄少75%。Advocate 1和Advocate 2均达到了主要终点,Lebrikizumab队列的IGA结果为43.1% (n = 283),⽽ Advocate 1的安慰剂队列 (n = 141) 为12.7%,Lebrikizumab为33.2% Advocate 2组 (n = 281) 的这一比例为 10.8%,⽽安慰剂组 (n = 146) 的比例为10.8%。第三项临床3期研究 Adhere(NCT04250337)是一项为期16周的随机、双盲、平⾏组研究,调查了Lebrikizumab联合局部皮质类固醇治疗211 名AD患者的疗效。患者按照2:1的比例随机分配,接受500 mg初始负荷剂量后的250 mg SC Lebrikizumab Q2W 治疗或安慰剂联合局部类固醇,中效(0.1% 曲安西龙丙酮乳膏)或低效类固醇(1%氢化可的松乳膏)。16周后,41.2%的 Lebrikizumab队列的IGA达到0或1,较基线降低2点或更多点,⽽安慰剂队列的这一比例为22.1%,早在第8周就达到了统计学显着性。达到EASI-75缓解的患者比例也显着增加。
托莱西单抗Tafolecimab (信达生物 Innovent Biologics, Inc.)
Tafolecimab (SINTBILO®)是一种⼈IgG2κ抗体,靶向前蛋⽩转化酶枯草杆菌蛋⽩酶/kexin 9型 (PCSK9)。2023年8⽉,信达⽣物宣布NMPA批准Tafolecimab⽤于治疗成⼈原发性⾼胆固醇⾎症和混合性⾎脂异常。SINTBILO®获批的给药⽅案包括150 mg Q2W、450mg Q4W和600 mg Q4W,所有这些方案都在临床3期研究中显示可有效降低低密度脂蛋⽩胆固醇(LDL-C)、总胆固醇(TC)、非⾼密度脂蛋⽩胆固醇(non-HDL-C)、载脂蛋⽩B(ApoB)和脂蛋⽩a(Lp(a))。
NMPA的批准基于三项安慰剂对照的临床3期临床研究CREDIT-1 (NCT04289285)、CREDIT-2 (NCT04179669)和CREDIT-4 (NCT04709536)的结果。CREDIT-1和CREDIT-2研究分别评估了Tafolecimab在中国非家族性⾼胆固醇⾎症和杂合⼦家族性⾼胆固醇⾎症受试者中的疗效和安全性。CREDIT-4研究包括患有任一类型⾼胆固醇⾎症的患者。接受Tafolecimab治疗的患者皮下注射450 mg Q4W或600 mg Q6W,持续48周 (CREDIT-1),150 mg Q2W 或450 mg Q4W,持续24周 (CREDIT-2),或 450 mg Q4W,持续 12 周 (CREDIT-4);在每项研究中,与安慰剂相比,Tafolecimab在接受该药物的患者中均显示出良好的安全性和显着的降脂功效。
纳鲁索拜单抗Narlumosbart(⽯药集团有限公司 CSPC Pharmaceutical Group Limited)
Narlumosbart(Jinlitai,津⽴泰)是⽯药集团⼦公司上海津曼特⽣物科技有限公司开发的一种⼈源化IgG4k单克隆抗体靶向核因⼦kB配体(RANK-L)受体激活剂。2023年9⽉,⽯药集团宣布NMPA批准Narlumosbart⽤于治疗⽆法切除或⼿术切除可能导致严重发病率的骨巨细胞瘤(GCTB)。
NMPA对Narlumosbart的批准基于两项临床研究的数据,即单臂、开放标签关键临床2期JMT103CN03研究(NCT04255576)和观察性JMT103CN03-1
Real-World研究(NCT05402865)。临床2期研究招募了139名参与者,符合条件的患者每4周皮下注射2 mg/kg Narlumosbart,并在治疗前4周的第8天和第15天接受负荷剂量。该观察性研究作为关键临床2期研究的对照,评估了抗RANK-L Denosumab和非Denosumab疗法在治疗⽆法⼿术挽救或严重术后并发症的中国⼈群中的疗效和安全性。研究结果显示,接受Narlumosbart治疗的患者临床疗效更好,肿瘤缓解率为93.5%,且有⾼于Denosumab组的趋势。
Narlumosbart还在一项多中⼼、随机、双盲、主动对照临床3 期研究(NCT05813665、CTR20231025)中进⾏评估,该研究对估计146 名患有不可切除或⼿术困难的GCTB患者进⾏了评估。患者将接受Narlumosbart 120 mg 或Denosumab 120 mg SC Q4W。该研究的主要结果指标是肿瘤缓解患者的百分比。
⽬前还在一项随机安慰剂/阳性对照临床2期研究 (NCT05278338) 中评估 Narlumosbart在骨质疏松症⽅面的疗效和安全性,该研究招募了约200名患有骨质疏松症的绝经后妇⼥。参与者将被随机分配接受60或90 mg Narlumosbart、60 mg Denosumab或安慰剂。所有治疗⼲预措施将在治疗阶段开始时和初始剂量后6个⽉内以单次皮下注射的形式进⾏。主要终点是治疗12个⽉时腰椎骨矿物质密度相对于基线的变化率。预计主要完成⽇期为2023年12⽉。
安瑞昔单抗Zuberitamab (浙江博锐生物制药有限公司 BioRay Pharmaceutical Co., Ltd.)
Zuberitamab (HS006, 安瑞昔®) 是一种⼈-⼩⿏嵌合IgG1κ抗CD20单克隆抗体,通过抗体依赖性细胞介导的细胞毒性(ADCC)和补体介导的细胞毒性杀死 B细胞。2023年5⽉12⽇,Zuberitamab获得中国NMPA批准⽤于治疗CD20 阳性DLBCL,这是成⼈中最常⻅的非霍奇⾦淋巴瘤(NHL)类型之一。在中国,DLBCL估计为约占所有NHL病例的40%。
NMPA 的批准是基于博锐生物进⾏的临床3期研究的结果。在这个多中⼼、随机、在中国开展的一项非劣效性试验(REFLECT)中,⼊组了487名患者,并接受了6个周期的Zuberitamab + CHOP或利妥昔单抗 + CHOP治疗。结果显示,与利妥昔单抗+CHOP组相比,接受Zuberitamab + CHOP治疗的个体表现出更⾼的CR率(85.66% vs 77.34%,P = 0.0378)。
此外,Zuberitamab组还表现出优于利妥昔单抗组的PFS率(3 年PFS率78.03% vs 70.90%)和总⽣存(OS)率(3 年OS率87.70% vs 83.14%)。特别是,对于⽣发中⼼B细胞亚型的患者,与利妥昔单抗组相比,Zuberitamab表现出显着更好的CR率、⽆事件⽣存率和OS率。此外,接受Zuberitamab治疗的患者的安全性与接受利妥昔单抗治疗的患者没有显着差异。
阿得贝利单抗Adebrelimab (江苏恒瑞医药股份有限公司Jiangsu Hengrui Medicine Co., Ltd.)
阿得贝利单抗(艾瑞利Arelili®)是江苏恒瑞医药股份有限公司研发的⼈源化抗PD-L1 IgG4κ抗体。2023年3⽉,该公司宣布,NMPA批准Adebrelimab联合化疗作为⼴泛性一线治疗⼩细胞肺癌(SCLC)。
NMPA批准Adebrelimab上市是基于在中国47个研究中⼼进⾏的安慰剂对照临床3期CAPSTONE-1研究(NCT03711305)的结果。符合条件的SCLC患者 (n = 462) 被随机分配(1:1)接受四到六个周期的carboplatin和etoposide联合Adebrelimab(20 mg/kg,每个周期的第1天)或匹配的安慰剂,然后接受Adebrelimab或安慰剂维持治疗。研究显示,与安慰剂联合化疗相比,Adebrelimab联合化疗显着改善了患者的OS(中位OS为15.3个⽉(95% CI 13.2-17.5) vs 12.8个⽉(11.3-13.7))。12 个⽉时的OS比率为62.9% vs 52.0%,24 个⽉时的OS比率为 31.3% vs 17.2%。根据独⽴审查委员会(IRC) 的规定,Adebrelimab + 化疗组的PFS为5.8个⽉(95% CI 5.6-6.9),⽽安慰剂 + 化疗组的PFS为5.6个⽉ (95% CI 5.5-5.7)(HR 0.67, 95% CI 0.54-0.83);6 个⽉时的PFS率为49.4% vs 37.3%,12个⽉时的PFS率为19.7% vs 5.9%。
正在对Adebrelimab与SCLC患者的化疗联合治疗相结合的其他肺癌临床3期研究进⾏评估(NCT04691063);与化疗联合作为可切除II期或III期NSCLC的围⼿术期治疗(NCT04316364),以及其他实体瘤患者的早期临床研究。
Divozilimab (BIOCAD)
Divozilimab (BCD-132, Ivlizi®) 是由BIOCAD开发的一种非岩藻糖修饰、⼈源化抗CD20 IgG1κ单克隆抗体。俄罗斯联邦卫⽣部于2023年3⽉批准Divozilimab ⽤于治疗多发性硬化症。多发性硬化症是一种⾃⾝免疫性疾病,免疫系统攻击髓鞘并扰乱神经信号的正常传输。Divozilimab结合B细胞上的CD20受体并导致这些细胞的耗竭。随着B细胞数量的减少,可以抑制炎症过程,从⽽有可能减缓疾病的进展并降低复发的频率和严重程度。
Divozilimab每6个⽉静脉注射一次。
临床3期MIRANTIBUS研究 (NCT05385744)使⽤活性参考药物(特⽴氟胺)评估了Divozilimab治疗复发性多发性硬化症患者的疗效和安全性。为了评估长期疗效和安全性,2期和3期研究的患者继续长期接受Divozilimab 3期研究和扩展研究。BIOCAD还在两项临床3期研究(分别为NCT05726630和NCT05730699)中评估Divozilimab在系统性硬化症(硬皮病)和视神经脊髓炎谱系疾病治疗中的作⽤。
 在线客服
在线客服








